醫藥科技有限公司")
取消
清空記錄
歷史記錄
清空記錄
歷史記錄


干貨分享 | 日本醫療器械分類及醫療器械主文檔
根據日本《藥品和醫療器械等產品質量、功效和安全保障法》(以下簡稱PMD Act):醫療器械是指用于診斷、治療或預防人類或動物疾病,或旨在影響人類或動物身體結構或功能(再生醫學產品除外)的器具或儀器等。
日本有兩個監管機構負責醫療器械的監管:厚生勞動省(MHLW)和PMDA。厚生勞動省負責行政行為,例如根據日本PMD Act來判斷產品是否被視為醫療器械以及負責指導或決定產品審批。PMDA則進行產品審查和上市后安全管理。
根據風險等級,日本將醫療器械分為四類[1]:I類(極低風險)、II類(低風險)、III類(中等風險)和IV類(高風險)。為使醫療器械能順利在日本上市,外國制造商必須根據PMD Act,通過日本上市許可持有人(MAH)或外國制造商指定的日本代理人以獲得批準/認證或提交備案。
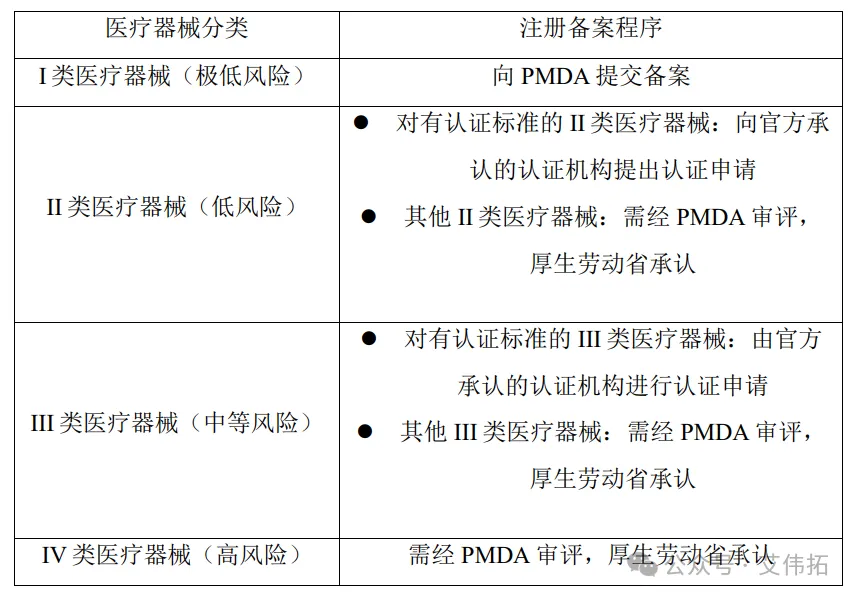
針對醫療器械原材料的登記備案,日本厚生勞動省專門制定了針對主文檔制度的指導文件《原料藥等注冊原簿引用指南》,以保護主文檔持有人的知識產權,同時提高審查效率。日本主文檔管理(Master File System,MF制度)的適用對象包括藥品原料藥、中間體和制劑原料;新輔料以及與以往配方比例不同的預混輔料;醫療器械原材料;再生醫療等產品原材料;容器、包裝材料[2]。
MF登記是一種自愿行為。MF申請者將申報資料遞交給PMDA官方,遞交后PMDA只進行形式審查。只有當醫療器械廠家在進行注冊引用該主文檔時,PMDA才會進行技術審評,具體請參照以前發布在AVT公眾號上的文章 “日本MF登記制度”。
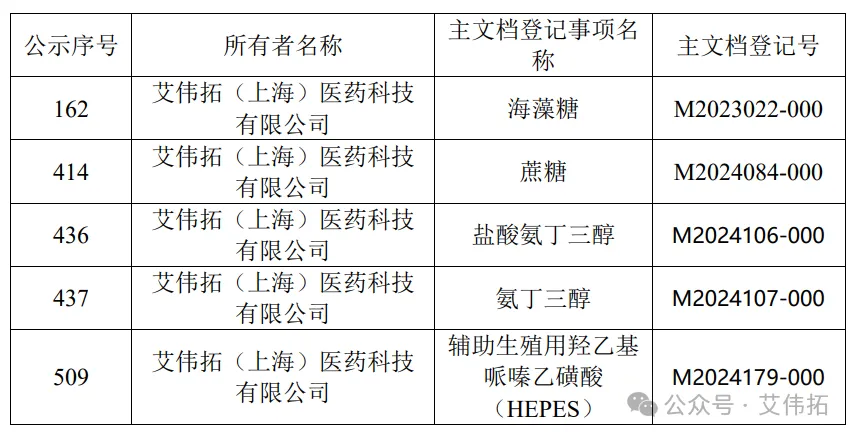
PMDA將在其官網上公布主文檔登記信息主文檔列表,包括MF登記號、名稱、登記日期、MF持有人的名稱和地址。
[1] Regulations and Approval/Certification of Medical Devices | Pharmaceuticals and Medical Devices Agency (pmda.go.jp)
[2] 《原料藥等注冊原簿引用指南》
艾偉拓醫療器械主文檔
(截至2024年6月25日)
 瀏覽器自帶分享功能也很好用哦~
瀏覽器自帶分享功能也很好用哦~



醫藥科技有限公司")

